Un fármaco existente puede prevenir la ruptura de la aorta.
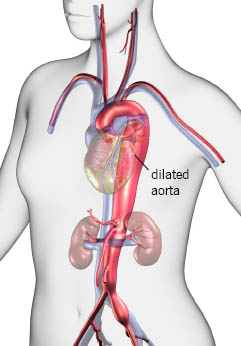
Cuando Hal Dietz llegó a la Universidad Johns Hopkins, en Baltimore, Maryland, en los 80, se obsesionó con la idea de ayudar a los niños que padecían el síndrome de Marfan, un trastorno poco común y a menudo fatal que puede provocar que la aorta, el vaso sanguíneo grande que lleva sangre del corazón, crezca tanto que se reviente.
“Nada de lo que hacíamos parecía hacer una diferencia en sus vidas”, comentó.
Dietz decidió enfocar su investigación en el gen que era la causa del síndrome de Marfan. Eso llevó a descubrimientos sorprendentes y a la prueba clínica, próxima a publicarse, de un fármaco que podría ayudar a quienes padecen este trastorno.
La labor de Dietz también inspiró investigación que podría resultar en una prueba de sangre para detectar aortas dilatadas, lo que posiblemente salvaría miles de vidas. La esperanza es que la prueba permita a los médicos operar antes de que se reviente la aorta, o identificar rápidamente una ruptura en la misma.
Los pacientes cuya aorta está en proceso de desintegrarse “necesitan tratamiento inmediato”, aseveró Scott A. LeMaire, profesor de cirugía y de fisiología molecular y biofísica en el Colegio Baylor de Medicina, en Houston.
Descubrir la mutación genética que causa el síndrome de Marfan solía ser un proceso lento y frustrante: aún no se habían inventado las máquinas de secuenciación que hoy son utilizadas para trazar rápidamente un mapa del ADN. Los investigadores tenían que examinar cada uno de los genes en regiones extensas de ADN.
La mutación fue descubierta en 1990, en la fibrilina 1, una proteína en el tejido conjuntivo, lo que indicaba que el tejido se deshacía porque sus “remaches” moleculares no funcionaban. De ser así, externó Dietz, “no había nada que se pudiera hacer para alterar el curso de la enfermedad”.
Sin embargo, la hipótesis de los remaches no explicaba algunas de las características más notables del Marfan en los niños: huesos notoriamente largos de brazos, piernas y dedos, ojos hundidos e inclinados hacia abajo, pómulos planos, mentones pequeños, masa muscular extremadamente baja y poca grasa corporal.
Hace unos 10 años, Dietz y sus colegas hallaron la respuesta en otra proteína, TGF- beta, o factor de crecimiento transformante beta, que dicta el comportamiento de las células durante el desarrollo y es utilizada en la reparación de heridas.
La función de la proteína depende de la fibrilina 1, la mismísima proteína que se ve alterada en el síndrome de Marfan. Normalmente, la fibrilina 1 conecta a la TGF-beta con el tejido conjuntivo. Sin embargo, descubrieron los investigadores, en un paciente con Marfan la fibrilina 1 es defectuosa, y el proceso se trastoca. En vez de unirse al tejido conjuntivo, la TGFbeta se aleja del mismo. Al flotar libremente en el torrente sanguíneo, provoca una conducta anormal en las células, lo que resulta en muchos de los problemas causados por el síndrome de Marfan.
Dietz buscó una manera de bloquear la función de la TGF-beta y descubrió que el losartán, un fármaco ampliamente utilizado para controlar la presión arterial, hacía precisamente eso.
En 2006, Kari Dostalik, de Urbandale, Iowa, cuya hija, Haley, padece el trastorno, acudió a una ponencia de Dietz en la conferencia anual sobre el síndrome de Marfan. Dietz mostró una diapositiva de un niño que sufría una forma severa del síndrome y a quien se le suministró losartán fuera de la prueba clínica. Dostalik había conocido al pequeño y a su familia en una conferencia anterior.
Antes del tratamiento, el niño lucía débil y cansado, pero tras tomar el medicamento “tenía una sonrisa de oreja a oreja”, narró ella.
Aún mejor, tan pronto como él y otros niños tomaron el fármaco, sus aortas dejaron de crecer; el losartán parecía revertir algunos de los efectos de la enfermedad.
Dos años después, Dietz y sus colegas publicaron datos en The New England Journal of Medicine sobre 17 niños severamente afectados a quienes se les dio el medicamento fuera de la prueba clínica. Antes de tomarlo, sus aortas crecían unos 3.5 milímetros al año; después, el índice se redujo a medio milímetro al año.
Los hallazgos sobre el Marfan empiezan a tener un efecto en un grupo más numeroso: quienes no padecen el trastorno, pero cuyas aortas se han dilatado y presentado una ruptura, una urgencia médica que pone en riesgo la vida. Parece que esos pacientes podrían tener las mismas señales moleculares delatoras que los pacientes de Marfan.
Amy Speck, de Knoxville, Maryland, inscribió a su hijo, Daniel, en la prueba clínica de Dietz. El niño tenía 8 años cuando fue diagnosticado, luego de que su médico notó una curvatura en su espina dorsal.
Speck afirmó que el tratamiento provocó “cambios maravillosos —todo se empezó a estabilizar”. La aorta de Daniel “crecía astronómicamente”, dijo, y esa dilatación se desaceleró tanto que no sería un candidato a la prueba clínica si hoy tratara de ingresar en ella.
Daniel, ahora de 15 años, evoluciona bien. “Es realmente asombroso”, expresó su madre.
Fuente: Clarín – Supl. The New York Times